引用本文
张林, 易先鹏, 王广杰, 范心宇, 刘辉, 王雪松. 基于网格重构学习的染色体分类模型. 自动化学报, 2024, 50(10): 2013−2021 doi: 10.16383/j.aas.c210303
Zhang Lin, Yi Xian-Peng, Wang Guang-Jie, Fan Xin-Yu, Liu Hui, Wang Xue-Song. A grid reconstruction learning model for chromosome classification. Acta Automatica Sinica, 2024, 50(10): 2013−2021 doi: 10.16383/j.aas.c210303
http://www.aas.net.cn/cn/article/doi/10.16383/j.aas.c210303
关键词
核型分析,染色体分类,特征重构,网格化
摘要
染色体的分类是核型分析的重要任务之一. 因其柔软易弯曲, 且类间差异小、类内差异大等特点, 其精准分类仍然是一个具有挑战性的难题. 对此, 提出一种基于网格重构学习(Grid reconstruction learning, GRiCoL)的染色体分类模型. 该模型首先将染色体图像网格化, 提取局部分类特征; 然后通过重构网络对全局特征进行二次提取; 最后完成分类. 相比于现有几种先进方法, GRiCoL同时兼顾局部和全局特征提取更有效的分类特征, 有效改善染色体弯曲导致的分类性能下降, 参数规模合理. 通过基于G带、荧光原位杂交 (Fluorescence in situ hybridization, FISH)、Q带染色体公开数据集的实验表明: GRiCoL能够更好地弱化染色体弯曲带来的影响, 在不同数据集上的分类准确度均优于现有分类方法.
文章导读
人体健康体细胞内有23对染色体, 包括22对常染色体和一对性染色体. 核型分析通常对分裂中期的染色体进行扫描、拍摄、处理、分割后, 获得染色体核型图用于染色体的分类及异常识别, 为染色体变异相关疾病的诊断及未知基因型疾病的发现提供有力参考. 因此, 核型分析已广泛应用于体外胚胎的细胞遗传学分析、产前诊断以及遗传病诊断. 但目前其分割、分类、识别等各个步骤均严重依赖核型专家的人工分析, 耗时耗力且依赖于经验. 因此, 不少学者开始研究相关的自动方法.
作为核型分析的核心环节, 染色体分类一直是核型分析领域的研究重点. 人类正常体细胞内不同类别的染色体形态相似, 但细节纹理部分差异显著; 且由于染色体的非刚性特点, 各染色体长短臂可以呈现出不同的弯曲状态. 针对核型图开展的染色体分类识别工作经历了传统方法[1-4]和基于卷积神经网络(Convolutional neural network, CNN)[5]方法两大发展阶段. 传统的染色体的自动分类方法一般由3个步骤构成[6-8], 包括提取染色体中心轴、根据中心轴提取分类特征以及设计分类器[9]. 其中, 染色体中心轴的提取取决于染色体的弯曲程度, 并决定后续的特征提取及处理. 因此传统方法在处理弯曲染色体时, 往往先作拉直处理, 再提取中心轴. 传统算法的各个环节具备较高的可解释性, 但算法整体环节多、复杂度高, 分类效果严重依赖中心轴的提取. CNN的提出为图像处理等领域开辟了新的天地[10-12], 设计合理的CNN模型能够自主地挖掘数据中的有用特征, 完成各类复杂任务, 并陆续应用于染色体图像的处理. 如Sharma等[13]、Swati等[14]提出了基于CNN的提取染色体特征分类方法. Sharma等[13]针对不同染色体的弯曲点和弯曲程度不同的问题, 先用传统方法[15]将染色体拉直, 然后再送入到卷积神经网络里进行分类, 对比是否进行拉直处理的实验分别获得68.5%和86.7%的分类准确率. Swati等[14]同样认为染色体弯曲给分类带来极大的困难. 因此采用提取中心轴和众包的方式拉直染色体, 然后再送入到孪生网络中进行染色体分类, 未拉直与拉直的准确率分别为68.5%与85.5%. 这些方法未能端到端的完成任务, 且准确率远远未达到要求. 而Zhang等[16]提出了一种基于利用改进的高分辨率网络模型预测弯曲染色体节点并分类的多任务方法. 该方法获得了98.1%的准确率, 但是需要预先标注弯曲节点, 花费大量人工成本. Qin等[17]提出的方法不做染色体拉直的预处理, 直接在高达87 831幅手工标注的G带图上使用Variafocal-Net模型, 可获得98.9%的准确度. 该模型采用两个残差模型串联, 第一个残差模型训练分类的同时学习一个坐标, 用于从原图切分出一个局部, 这个局部再提取特征进行分类. 而该数据集仅残差网络(Residual neural network, ResNet)[18]就得到96.9%的准确度. 由于该模型没有针对染色体弯曲问题设计, 在面对无法提供海量训练数据的实际问题时, 较难达到预期的性能. 为应对数据量少的情况, Wu等[19]提出应用生成对抗网络生成染色体图像, 然后再送入到CNN中进行分类. 由于生成的数据和真实数据仍有区别, 效果提升有限, 其在5474幅图像上的准确率为58.9%, 扩充250倍后可提升至63.5%. 染色体弯曲问题可通过大量的数据集来解决, 但是大数据集标注代价高昂. 因此, 针对小规模染色体数据集, 开发一种有效提升染色体识别性能的分类方法非常重要.
染色体核型图的分类、识别任务具有类间差距小、类内差距大的特点, 因此其分类任务可借鉴自然图像的细粒度分类任务[5,20]. 目前细粒度分类任务多通过仅基于图像级标签实现的弱监督分类和带有边界框、零件关键点等先验信息的强监督分类两种思路实现[21]. 在弱监督分类算法方面, 基于显式特征映射的池化框架利用核函数能够较好地捕捉CNN提取特征间的高阶信息, 提升细粒度分类精度, 但局部特征的提取方法仍有待改善[22]. 基于两个CNN特征抽取器建立的双线性模型框架以平移不变的方式对局部成对特征进行提取, 获得的特征更精细, 但特征之间较难建立有效的关联, 且模型参数量太大, 不利于模型的实际应用[23]. 另有一些破坏与重构学习模型和循环注意力卷积神经网络模型[24-25], 通过强化局部特征对分类任务的贡献, 更好地习得具有判别性的特征, 提高分类的准确性, 但也未能充分建立局部特征间的有效关联. 而在强监督分类方面, Wei等[26]建立全卷积网络结构, 基于细粒度图像的局部标注定位有区别的部分, 并生成目标及对应掩码, 提取辨别性较高的特征. 在此基础上, 进一步搭建四路掩膜CNN模型, 聚合提取出的特征, 构建高精度分类器. 但强监督分类需额外标注, 代价太大.
综上所述, 已有的染色体分类方法和细粒度分类思想在训练样本的数量、质量及模型对图像的局部特征提取能力、局部特征的综合利用程度、计算量和最终准确度方面都存在一定的局限. 为此, 本文针对染色体弯曲及类内差距大、类间差距小导致的分类难题, 基于微分思想建立网格重构学习(Grid reconstruction learning, GRiCoL)模型提取细粒度局部特征, 在不增加额外标注要求前提下有效提高分类性能. 结果表明, GRiCoL可有效提高染色体分类精度, 在G带、荧光原位杂交(Fluorescence in situ hybridization, FISH)、Q带三个公共图像库上的分类准确率分别达到0.995, 0.973和0.972.
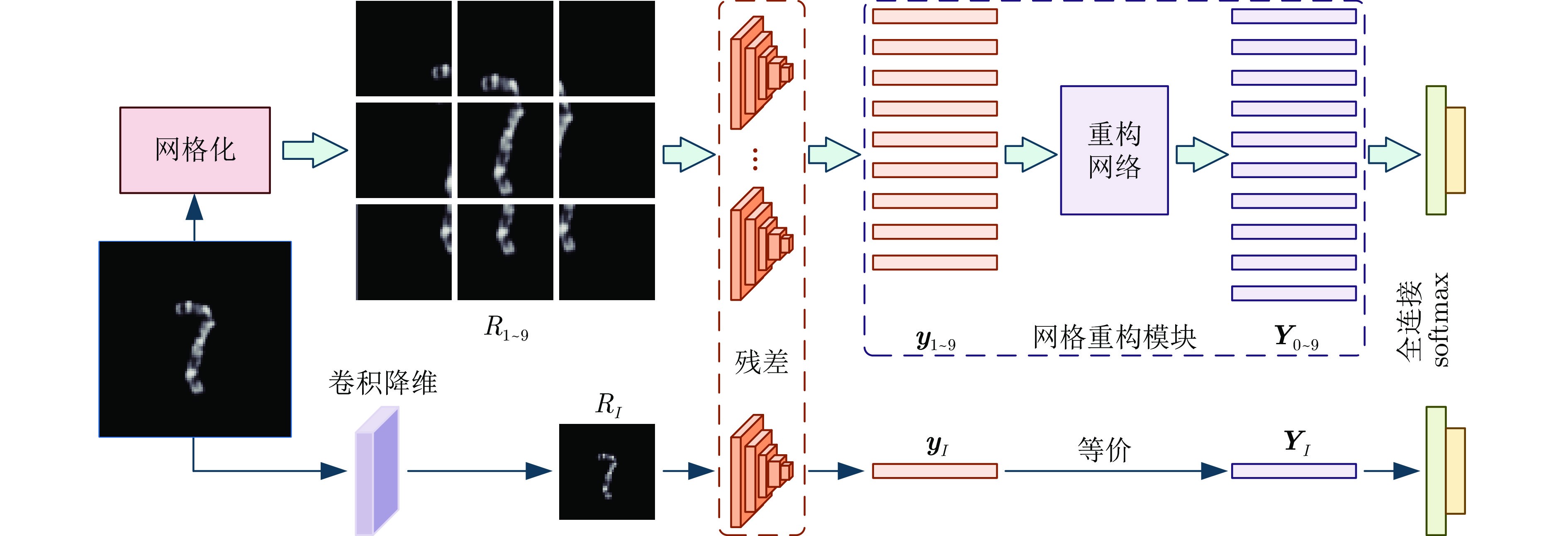
图1基于网格重构学习的染色体分类模型
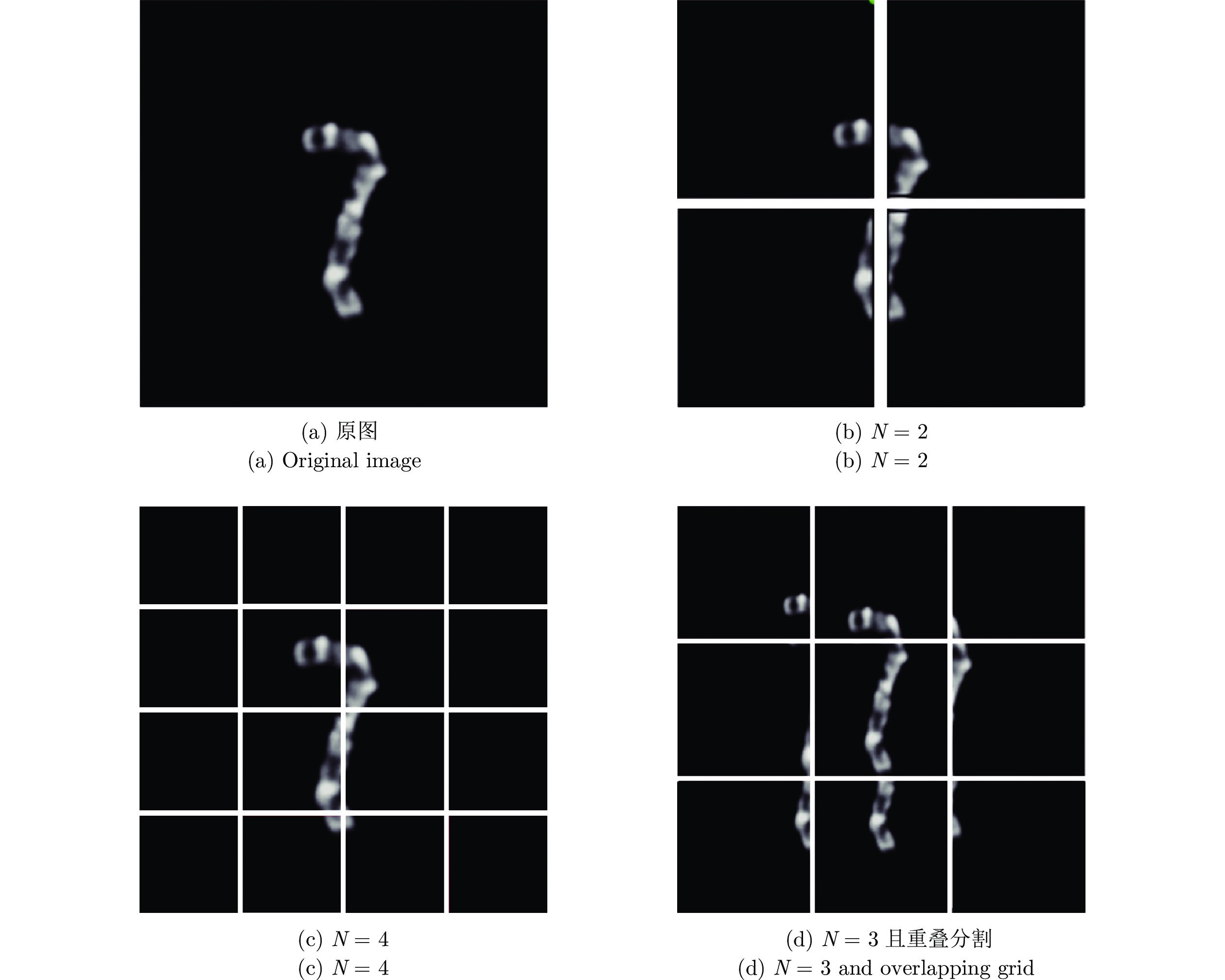
图2染色体图像网格化效果
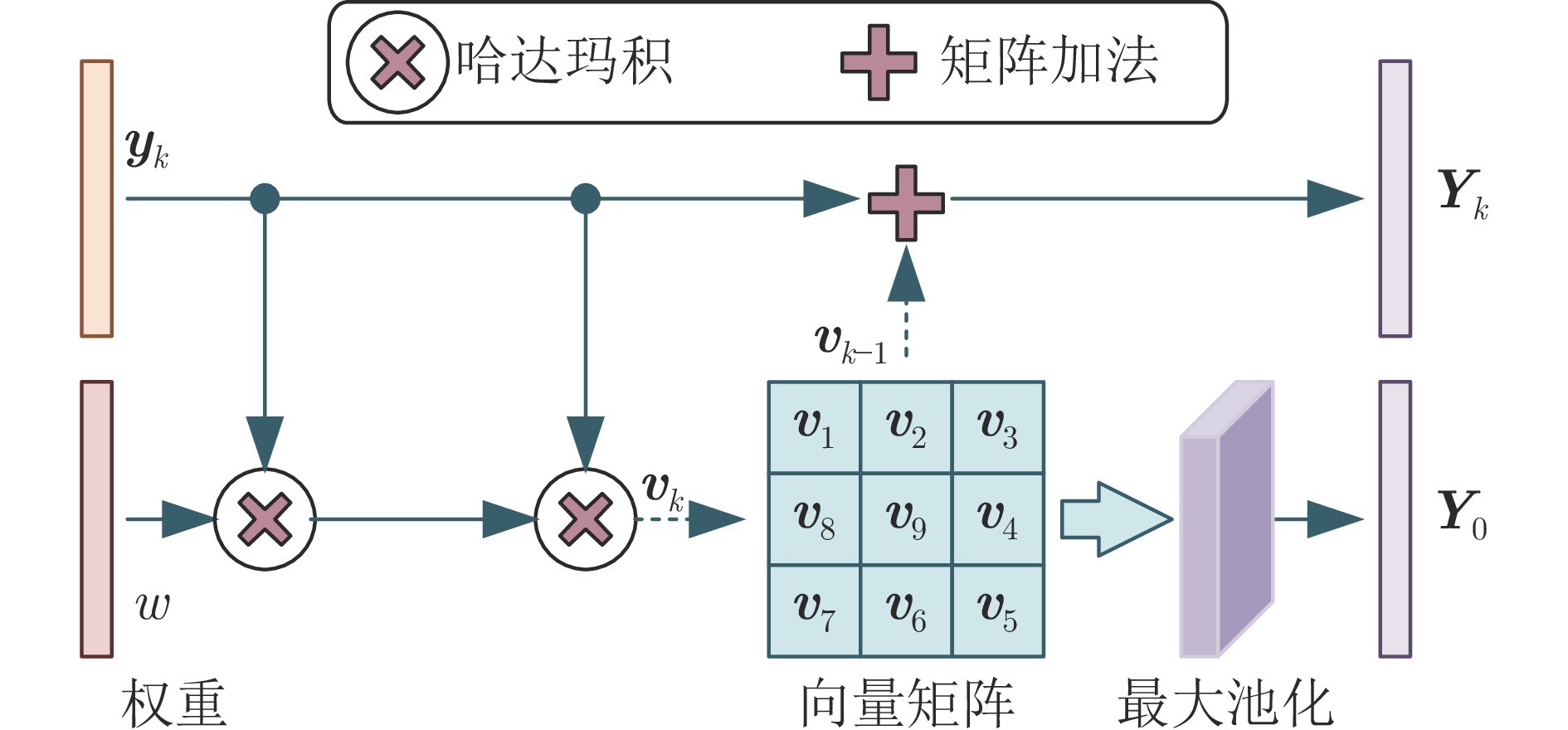
图3重构网络模型
染色体由于其柔性易弯曲的特点, 目前其自动分类存在较大困难. 为解决该问题, 本文提出GRiCoL模型, 利用微分思想对染色体图像进行合理切分, 弱化弯曲的影响, 通过残差网络、重构网络等模块提取特征后, 完成分类模型的构建. 本文分别基于G带图、FISH图和Q带图三个染色体公共图像库对GRiCoL模型进行训练和评估, 对比实验结果显示, 本文提出的GRiCoL能够有效提高染色体图像的分类准确率, 且表现出更强的泛化能力及对不同显带成像技术的适应性. 此外, GRiCoL模型可以在较小的数据集上完成有效的训练, 所提取的特征也呈现出较好的解释性. 因此, 本文所提出的GRiCoL网络模型能够较好地解决弯曲染色体的分类问题, 并为染色体的端到端自动核型分析提供新的思路.
作者简介
张林
中国矿业大学信息与控制工程学院教授. 主要研究方向为生物信息学, 医学图像处理, 机器学习. E-mail:[email protected]
易先鹏
中国矿业大学信息与控制工程学院硕士研究生. 主要研究方向为医学图像处理. E-mail:[email protected]
王广杰
中国矿业大学信息与控制工程学院硕士研究生. 主要研究方向为医学图像处理. E-mail:[email protected]
范心宇
中国矿业大学信息与控制工程学院博士研究生. 主要研究方向为图像处理. E-mail:[email protected]
刘辉
中国矿业大学信息与控制工程学院副教授. 主要研究方向为生物信息学, 医学图像处理, 机器学习. E-mail:[email protected]
王雪松
中国矿业大学信息与控制工程学院教授. 主要研究方向为人工智能, 机器学习. 本文通信作者. E-mail:[email protected]